
The biggest success of AI in science so far is the AlphaFold 2 system. This is a deep learning system which has made large strides on a fundamental scientific problem: how to predict the 3-dimensional structure of a protein from the sequence of amino acids making up that protein. This breakthrough has helped set off an ongoing deep learning revolution in molecular biology. While obviously of interest to molecular biologists, I believe this is of much broader interest for science as a whole, as a concrete prototype for how artificial intelligence may impact discovery. In this short survey talk I briefly discuss questions including: how can such systems be validated? Can they be used to identify general principles that human scientists can learn from? And what should we expect a good theory or explanation to provide anyway? The focus of the talk is extant results and the near term, not the longer-term future. However, I hope it may help in grounding an understanding of that longer-term future, and of the benefits and risks of AI systems.
The text for a talk given at the Metascience 2023 Conference in Washington, D.C., May 2023.
In 2020 the deep learning system AlphaFold 21 surprised biologists when it was shown to routinely make correct near atomic-precision predictions for protein structure. That is, using just the linear sequence of amino acids making up a protein, AlphaFold was able to predict the positions of the atoms in the protein. These results were not cherrypicked, but rather emerged from an adversarial, blind competition with over a hundred other modeling groups. In a few cases, AlphaFold2 has even exceeded experimental accuracy, causing existing experimental results to be re-evaluated and improved.
While AlphaFold is impressive, it's also far from complete: it's really a bridge to a new era, opening up many scientific and metascientific questions. These include: what we expect a good theory or explanation to provide; what it means to validate that understanding; and what we humans can learn from these systems. Most of all: whether and how AI systems may impact the progress of science as a whole, as a systemic intervention. In my talk, I'll treat AlphaFold as a concrete prototype for how AI may be used across science. For these reasons, it's valuable for metascientists to engage with AlphaFold and successor systems, even if you have no prior interest in proteins or even in biology.
The talk is a survey. I am not a molecular biologist, so my apologies for any errors on that front. That said, it's been enjoyable and often inspiring to learn about proteins. Let me take a few minutes to remind you of some background for those of you (like me) who aren't biologists. Molecular biology feels a bit like wandering into an immense workshop full of wonderful and varied machines. Large databases like UniProt contain the amino acid sequences for hundreds of millions of proteins. You have, for example, kinesin proteins, which transport large molecules around the interior of the cell. You have haemoglobin, which carries oxygen in your blood, and helps power your metabolism. You have green fluorescent protein, which emits green light when exposed to ultraviolet light, and which can be used to tag and track other biomolecules. All these and many more molecular machines, created and sorted by the demanding sieve of evolution by natural selection. Every individual machine could be the subject of a lifetime's study. There are, for instance, thousands of papers about the kinesin superfamily, and yet we're really just beginning to understand it. But while this wealth of biological machines is astonishing, we don't a priori know what those machines do, or how they do it. We have no instruction manual, and we're trying to figure it out.
In fact, for the vast majority of the hundreds of millions of proteins known3, all we can easily directly determine is the basic blueprint: using genome sequencing we can find the linear sequence of amino acids that form the protein, at a cost of no more than cents. But proteins are tiny 3-dimensional structures, typically nanometers across, making it extremely difficult to directly image them. For a single protein it will routinely take months of work to experimentally determine the corresponding 3-dimensional structure, usually using x-ray crystallography or cryo-electron microscopy or NMR.
This discrepancy really matters. It matters because understanding the shape is crucial for understanding questions like:
Understanding the shape of a protein doesn't tell you everything about its function. But it is fundamental to understanding what a protein can do, and how it does it.
Ideally, we'd be able to determine the shape from the amino acid sequence alone. There are reasons chemists and biologists expect this to often be possible4, and in the 1970s, scientists began doing physics simulations to attempt to determine the shape a protein will fold into. Many techniques have been adopted since (kinetics, thermodynamics, evolutionary, ….) For a long time these made only very slow progress. And even for that progress, one must wonder if modelers are cherrypicking, even with the best will in the world. There were, for instance, claims in the 1990s from Johns Hopkins to have largely "solved" the protein structure prediction problem5.
To assess progress in a fair but demanding way, in 1994 a competition named CASP (Critical Assessment of protein Structure Prediction) was begun. Running every two years, CASP asks modelers to do blind predictions of protein structure. That is, they're asked to predict structures for proteins whose amino acid sequence is known, but where biologists are still working on experimentally determining the three-dimensional structure, and expect to know it shortly after the competition is over, so it can be used to score predictions. The score for any given model is (very roughly) what percentage of amino acids positions are predicted correctly, according to some demanding threshold6. As you can see, through the 2010s the winner would typically score in the range 30 to 507:
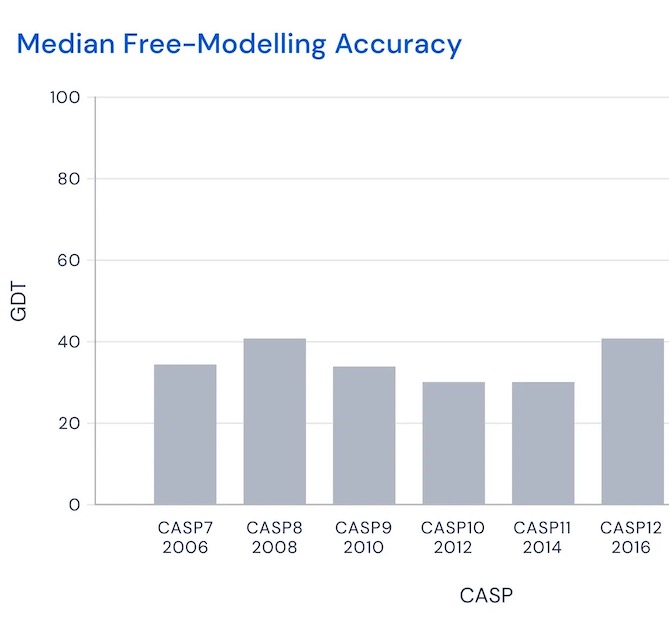
That is, the winner would (roughly) place somewhat than half of amino acids near perfectly. Then, in 2018 and 2020, DeepMind entered with AlphaFold and AlphaFold 2:
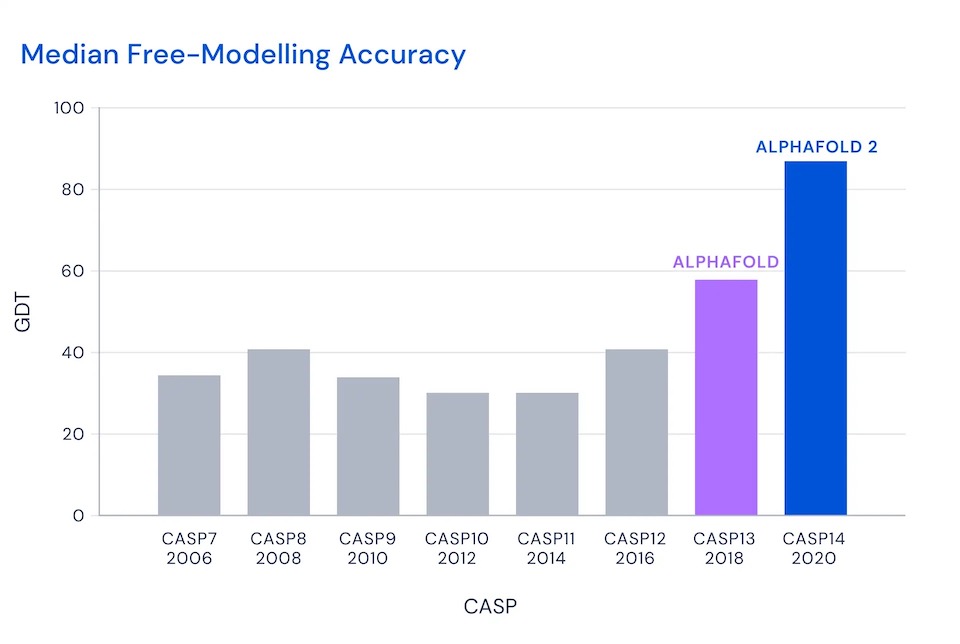
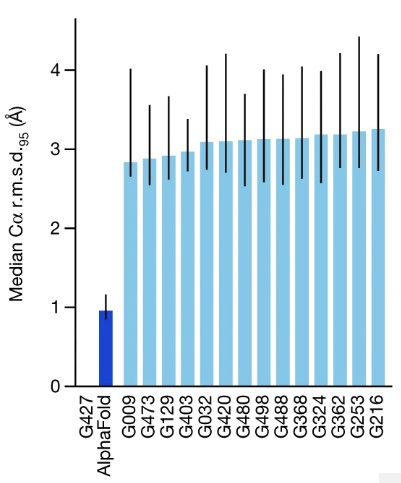
I'll focus on CASP 14 in 2020, when AlphaFold 2 achieved of score of 87, so roughly 87 percent of amino acids within a demanding threshold. This was AlphaFold's score in the most demanding category, the free modeling category, where contestants are asked to predict the structure of proteins for which few similar proteins were previously known. Across all categories, AlphaFold predicted the position of the alpha-carbon atoms in the backbone with a root mean square distance of 0.96 Angstroms8. The next-best technique was roughly one third as accurate, with a root mean square distance of 2.8 Angstroms9:
For comparison, the van der Waals diameter of a carbon atom is roughly 1.4 Angstroms. AlphaFold's accuracy is often comparable to experimental accuracy. The co-founder of CASP, John Moult of the University of Maryland, said10:
We have been stuck on this one problem – how do proteins fold up – for nearly 50 years. To see DeepMind produce a solution for this, having worked personally on this problem for so long and after so many stops and starts, wondering if we’d ever get there, is a very special moment.
That is a very strong statement, and it's worth digging into in what sense AlphaFold solves the protein structure prediction problem, and what remains to be understood. However, even AlphaFold's competitors were laudatory. Here's Mohammed AlQuraishi of Columbia University11:
Does this constitute a solution of the static protein structure prediction problem? I think so but there are all these wrinkles. Honest, thoughtful people can disagree here and it comes down to one’s definition of what the word “solution” really means… the bulk of the scientific problem is solved; what’s left now is execution.
That was two-and-a-half years ago. I think a reasonable broad view today is: AlphaFold is a huge leap, but much remains to be done even on the basic problem, and many enormous new problems can now be attacked.
AlphaFold's high-level architecture would take several hours to describe. I want to emphasize just a few points today:
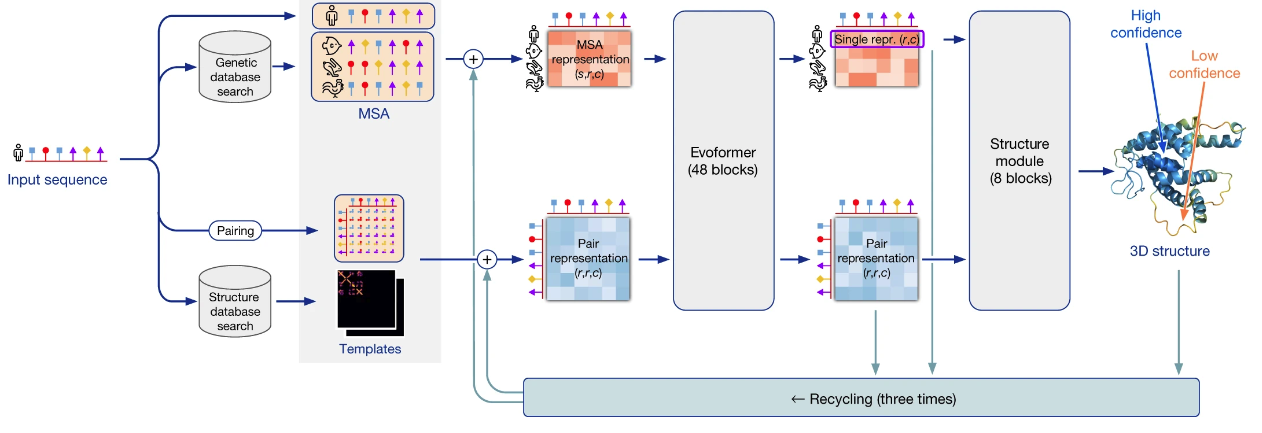
It's a deep neural network, meaning a hierarchical model, with 93 million parameters learned through training. An amino acid sequence is input, and a 3-dimensional structure is output, together with some error estimates quantifying how confident AlphaFold is in the placement of each amino acid. The basic training data is the protein data bank (PDB), humanity's repository of the protein structures experimentally determined since the 1970s. At training time, that was 170,000 protein structures (though a small fraction were omitted for technical reasons). The parameters in the AlphaFold network are adjusted using gradient descent to ensure the network outputs the correct structure, given the input.
That's a (very!) broad picture of how the network learned to predict structures. Many more ideas were important. One clever and important (albeit existing) idea was a way to learn from the hundreds of millions of known sequences for proteins. The idea is to find many other proteins whose amino acid sequences are similar to the input protein, meaning they're likely to be evolutionarily related. Maybe, for instance, it's essentially the same protein, but in some other species. AlphaFold tries to find many such similar proteins, and to learn from them. To get an intuition for how it might do that, suppose by looking at many similar proteins AlphaFold finds that there is some particular pair of amino acids which are a long way apart in the linear chain, but where changes in one amino acid seem to be correlated to changes in the other. If you saw that you might suspect these amino acids are likely to be close together in space, and are co-evolving together to preserve the shape of the protein. In fact, this often does happen, and it provides a way AlphaFold can learn both from known structural information (in the PDB), and from known evolutionary information (in protein sequence databases). A nice way of putting it, in a talk from John Jumper, lead author on the AlphaFold paper, is that the physics informs AlphaFold's understanding of the evolutionary history, and the evolutionary history informs AlphaFold's understanding of the physics.
You might wonder: is AlphaFold just memorizing its training data, or can it generalize? CASP provides a basic validation: the competition structures were not in the PDB at the time AlphaFold predicted them. Furthermore, CASP is in some sense a "natural" sample: the structural biology community prefers to solve structure which are biologically important12. So it suggests some ability to generalize to proteins of interest to biologists at large.
What if we use deep learning to study proteins which don't occur in nature, maybe as the result of mutations, or as part of protein design? There's a huge amount of work going on, and frankly it's an exciting mess right now, all over the place – any reasonable overview would cover dozens if not hundreds of papers. There are papers saying AlphaFold works badly for such-and-such a type of mutation, it works well for such-and-such a kind of mutation, all sorts of deep learning fixes for protein design, and so on. My takeaway: it's going to keep biologists busy for years figuring out the shortcomings, and improving the systems to address those shortcomings.
Some interesting tests of deep learning's ability to generalize were done by OpenFold13, an open source near-clone of AlphaFold. For instance, they retrained OpenFold with most broad classes of topology simply removed from the training set. To do this, they used a standard classification scheme, the CATH classification of protein topology. They removed 95% (!!!) of the topologies from the training set, and then retrained OpenFold from scratch. Even with the great majority of topologies removed, performance was similar to AlphaFold 114: just a couple of years prior to AlphaFold 2 it would have been state-of-the-art.
In another variation on this idea, they retrained OpenFold starting from much smaller subsamples of the protein data bank. Instead of using 170,000 structures, they retrained with training sets as small as 1,000 structures, chosen at random. Even with such a tiny training set, they achieved a performance comparable to (in fact, slightly better than) AlphaFold 1. And with just 10,000 structures as training data they achieved a performance comparable to the full OpenFold.
As a practical matter, the question of AlphaFold generalization matters in part because DeepMind has released AlphaFold DB, a database of 215 million protein structures, including the (almost-complete) proteomes of 48 species, including humans, mice, and fruit flies. These were obtained by taking genetic sequences in UniProt, and then using AlphaFold to predict the structure. You can view the whole process as:
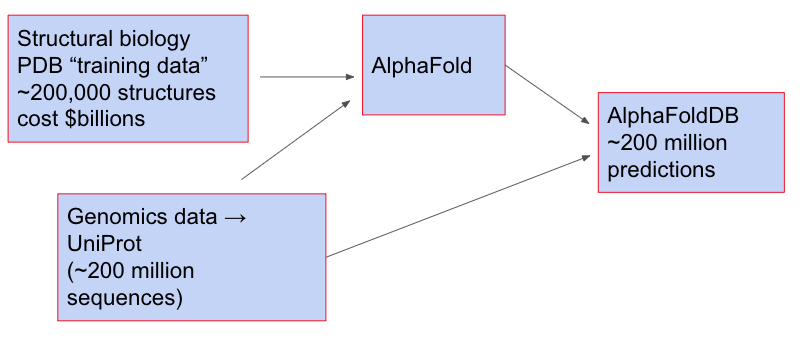
It's an astonishing act of generalization. If we had a perfectly reliable model, it would extend our understanding of protein structures by roughly three orders of magnitude. It's worth emphasizing that no additional experiments were done by AlphaFold; no additional data were taken. And yet by "just thinking" it was possible to obtain a very large number of predictions that people expect to be very good. I've heard from several biologists variations on the sentiment: "No-one would take an AlphaFold prediction as true on its own; but it’s an extremely helpful starting point, and can save you months of work".
As the models get better still, I expect the line between model and experiment to become blurry. That may sound strange, but in fact traditional "experimentally-determined structures" actually require immensely-complicated theories to go from data to structure. If you believe AlphaFold (or a successor) offered a stronger theory, you might end up believing the predictions from the deep learning system more than you believe (today's) "experimental results". There are early hints of this beginning to happen. In the CASP assessment, AlphaFold performed poorly on several structures which had been determined using NMR spectroscopy. A 2022 paper15 examined "904 human proteins with both Alpha-Fold and NMR structures" and concluded that "Alpha-Fold predictions are usually more accurate than NMR structures". One of the authors, Mike Williamson, actually helped pioneer the use of NMR for structural biology.
To make the same point in a simpler setting: how we interpret the images from a telescope depends upon our theory of optics; if we were to change or improve our theory of optics, our understanding of the so-called "raw data" from the telescope would change. Indeed, something related has really happened in our understanding of the bending of light by gravitation. In that case, we've changed our understanding of the way light travels through space, and that has affected the way we interpret experimental data, particularly in understanding phenomena like gravitational lensing of distant galaxies.
In a similar way, "experimental" protein structure determination depends strongly on theory. You can get a gist for this in x-ray crystallography, which requires many difficult steps: purification of protein sample; crystallization (!) of the proteins; x-ray diffraction to obtain two-dimensional images; procedures to invert and solve for the three-dimensional structure; criteria for when the inversion is good enough. A lot of theory is involved! Indeed, the inversion procedure typically involves starting with a good "guess", a candidate search structure. Often people use related proteins, but sometimes they can't find a good search structure, and this can prevent a solution being found. AlphaFold has been used to find good search structures to help invert data for particularly challenging structures. So there is already a blurry line between theory and experiment. I expect figuring out how to validate AI "solutions" to problems will be a major topic of scientific and metascientific interest in the years to come.
Any model with 93 million learned parameters is complicated. It's not a "theory" or "explanation" in the conventional sense. You might wonder: can AlphaFold (or a successor) be used to help discover such a theory, even if only partial? Might, for instance, a simple set of principles for protein structure prediction be possible? And what, exactly, is AlphaFold 2 learning? To avoid disappointment, let me say: we don't yet know the answers to such questions. But pursuing them is a useful intuition pump for thinking about the role of AI in science.
One approach to finding such principles is "behaviorist artificial psychology": inferring high-level principles by observing the behavior of the system. Of course, AlphaFold's detailed predictions have already been used widely by biologists, for things like discovering new binding sites on proteins. But while this is very useful, it isn't the same as inferring novel high-level principles about protein structure. However, there are other important deep learning systems in which interesting new high-level principles have been found by observing behavior.
For instance, by observing the AlphaZero chess system16 behaviors have been inferred which violated conventional chess grandmaster wisdom. These behaviors were announced in December 201817 and January 201918. A recent paper19 examined these behaviors, and tried to determine how (and whether) human players had changed in response to the system. They found few changes in the top ten players in 2019, with one exception: the world's top player then and now, Magnus Carlsen. They identify multiple ways in which Carlsen changed his play significantly in 2019, plausibly influenced by AlphaZero.
Among the changes: Carlsen advanced his h pawn early in the game20 much more frequently (a 333% increase with white, a 175% increase with black). He changed his opening strategies for both white and black. Indeed, his two most common 2019 opening strategies with white (variations of the Queen's Gambit declined and of the Grunfeld Defense) were openings he didn't play at all in 2018. And with black he played the Sicilian less than 10% of the time in 2018, but 45% of the time in 2019. They also observed an increase in Carlsen's willingness to sacrifice material. All these changes were consistent with learning from AlphaZero. Carlsen lost no classical games in 2019, and was the only top grandmaster to considerably improve his Elo rating, despite already being the top-rated player: he increased by 37 rating points. By contrast, 6 of the top 10 players actually lost points, and none of the other 3 gained more than 6 points. Of course, we do not know how much of this was due to Carlsen learning from AlphaZero, but the paper makes a plausible case that Carlsen learned much from AlphaZero.
Such behavioral observation is interesting but frustrating: it doesn't tell us why these behaviors occur. We may know that AlphaZero likes to advance the h pawn early, but what we'd really like is to understand why. Can we instead look inside the neural networks, and understand how they do what they do? As far as I know this kind of investigation has only been done casually for AlphaFold. But for simpler neural nets people are discovering interesting structure. Last year, for example, Neel Nanda and Tom Lieberum21 trained a single-layer transformer neural network to add two integers modulo 113. At first, the network simply memorized all the examples in the training set. It could add those well, but (unsurprisingly) gave terrible performance elsewhere. But as they trained the network for much longer, it transitioned to performing vastly better on held-out examples. Somehow, with no additional training data, it was learning to add.
Nanda and Lieberum spent weeks looking inside the network to understand what had changed. By reverse engineering the network they discovered it had learned to add in a remarkable way. The rough gist is: given numbers x and y the network computed a wave e^{2\pi i kx/113}, then did a phase shift to e^{2\pi i k x/ 113} \times e^{2\pi i ky/113}, and finally tried to find a canceling phase shift z which would result in the wave e^{2 \pi i k(x+y-z)/113} having the flattest possible "tone". It's a radio frequency engineer or group representation theorist's way of doing addition. This came as quite a surprise to Nanda and Lieberum. As Nanda said:
To emphasize, this algorithm was purely learned by gradient descent! I did not predict or understand this algorithm in advance and did nothing to encourage the model to learn this way of doing modular addition. I only discovered it by reverse engineering the weights.
This sequence of events greatly puzzled me when first I heard it: why did the network switch to this more general algorithm? After all, it initially memorized the training data, and provided excellent performance: why change? The answer is that during training the neural network pays a cost for more complex models: the loss function is chosen so gradient descent prefers lower-weight models. And the wave algorithm is actually lower-weight. It's a kind of mechanical implementation of Occam's razor, preferring an approach which is simpler and, in this case, more general. Indeed, by varying the loss function you can potentially impose Occam's razor in many different ways. It's intriguing to wonder: will one day we be able to do similar things for systems like AlphaFold, perhaps discovering new basic principles of protein structure?
I hope you've enjoyed this brief look at AlphaFold and reflections on the use of AI in science. Of course, there's far more to say. But I believe it's clear there are many fundamental scientific and metascientific questions posed by AI systems. Most of all: as they get better at different types of cognitive operation, and as they become more able to sense and actuate in the environment, how will they impact science as a whole? Will AI systems eventually systematically change the entire practice of science? Will they perhaps greatly speed up scientific discovery? And if so, what risks and benefits does that carry?
At the end of my talk many interesting questions were asked by audience members. One striking question came from Evan Miyazono. It was, roughly: "As a target for AI, protein structure prediction benefits from having a lot of very clean prior data which the system can learn from. How useful do you think AI will be for problems that don't have lots of clean prior data to learn from?"
It's a great question. A year or so ago I would have said that yes, it seems challenging to apply AI when you're working with messier problems with less clear metrics of success. But I've changed my mind over the year, as I've better understood foundation models, transfer learning, and zero-shot learning.
To explain why I've changed my mind, let me give two pieces of context. First, some informal gossip: many people have observed that ChatGPT is much better at coding than GitHub Copilot. I don't know the reason for sure. But a common speculation I've heard from informed people is that training on lots of text (as well as code) substantially improves the model's ability to code. Somehow, it seems the regularities in the text are improving ChatGPT's ability to understand code as well.
The second piece of context: a similar idea is used with AlphaFold. To explain this I need to explain a piece of AlphaFold's training that I didn't discuss in the body of the talk: it actually implicitly includes a large language model, treating the sequence of amino acids as a kind of "text" to be filled in. To understand this, recall that given a sequence of amino acids, AlphaFold looks up similar sequences in existing genetic databases. And during training, just like a language model, AlphaFold masks out (or mutates) some of the individual amino acids in those sequences, and attempts to predict the missing values. Doing this forces the network to learn more about the structure of the evolutionary information; it then takes advantage of that forced understanding to do better at protein structure prediction. In this way, AlphaFold benefits from transfer learning from a related domain (genetics) where far more information is available.
Returning to the broader context of science, I expect large multi-modal foundation models will: (a) gradually begin to outperform special-purpose systems, just as large language models often outperform more specialized natural language algorithms; and (b) they will exhibit zero-shot or few-shot learning. These large multi-modal models will be trained not just on text, but also on images and actions and code and genetic data and geographic data and all sorts of sensor and actuator data. And, just as in the language models, they will use what they understand in one domain to help inform their work in adjacent, often messier domains, which the model may perhaps have limited information about. Indeed, there are already hints of protein language models moving in this direction.
Of course, all this is merely a story and an intuition. It hasn't happened yet, except in very nascent ways, and I may be quite wrong! I certainly expect new ideas will be needed to make this work well. But hopefully that sums up how my intuition has changed over the last year: I think AI models will, in the next few years, be surprisingly good at addressing messy problems with only a little prior data. And they'll do it using multi-model foundation models, transfer learning, and zero- and few-shot learning.
This talk was supported by the Astera Institute. Thanks to Alexander Berger, David Chapman, Evan Miyazono, Neel Nanda, and Pranav Shah for comments that helped improve this talk. And thanks to my many correspondents on Twitter, who have helped me deepen my understanding of AI and of molecular biology.
For attribution in academic contexts, please cite this work as:
Michael Nielsen, "How AI is impacting science", https://michaelnotebook.com/mc2023/index.html, San Francisco (2023).
John Jumper, Richard Evans, Alexander Pritzel et al, Highly accurate protein structure prediction with AlphaFold, Nature (2021). See also this colab, which provides an executable version of AlphaFold from Google DeepMind, the company behind AlphaFold. Note that other sources provide faster implementations, with comparable performance. See, for instance this ColabFold colab. And, as we shall see later, other groups have developed open source implementations of the code training the system, not just for making structure predictions.↩︎
I'll use "AlphaFold" to refer to AlphaFold 2. Of course, it is a followup to an earlier AlphaFold system for protein structure prediction. However, we won't discuss that system in any depth, and so I will usually omit the number. Note that AlphaFold 1 and AlphaFold 2 had very different architectures, apart from both being deep learning systems for protein structure prediction.↩︎
Indeed, billions may be found (though of less clear provenance!) in metagenomics databases.↩︎
This is a complicated subject, sometimes referred to as Anfinsen's hypothesis or Anfinsen's dogma. It is not true all the time – there are certainly circumstances under which the same amino acid sequence can fold in different ways, or has no stable structure. And yet it has served as an extremely useful basis for beginning to understand proteins and their function. In this talk I'll mostly assume it's true.↩︎
See, for instance, this remarkable press release from Johns Hopkins, about the work of George Rose and Rajgopal Srinivasan "solving" the problem of predicting the backbone structure for globular proteins.↩︎
More precisely, the global distance test (GDT) used in CASP is an average of four different percentages, measuring the fraction of predictions for the backbone alpha carbon atoms which fall within 1, 2, 4, and 8 Angstroms of the experimental structures. For comparison, I am told that good experimental structures are often thought to be accurate to about 1-2 Angstroms; better has been achieved, but worse is not uncommon, depending on the technique.↩︎
This graph and the one that follow are adapted from AlphaFold: a solution to a 50-year-old grand challenge in biology (2021).↩︎
This was for 95% coverage of the alpha carbons. For 100% coverage, it was 1.5 Angstroms.↩︎
Figure from: John Jumper, Richard Evans, Alexander Pritzel et al, Highly accurate protein structure prediction with AlphaFold, Nature (2021).↩︎
Quoted in: AlphaFold: a solution to a 50-year-old grand challenge in biology (2021).↩︎
Mohammed AlQuraishi, AlphaFold2 @ CASP14: “It feels like one’s child has left home.” (2020).↩︎
Of course, other concerns like the experimental tractability of a structure also play a role.↩︎
Gustaf Ahdritz, Nazim Bouatta, Sachin Kadyan et al, OpenFold: Retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization (2022).↩︎
They use the local distance difference test (lDDT-C\alpha) as their metric, rather than the global distance test emphasized in AlphaFold's performance on CASP. I doubt this makes much difference, but haven't checked; I must admit, I wonder if there's some important point I'm missing here.↩︎
Nicholas J. Fowler and Mike P. Williamson, The accuracy of protein structures in solution determined by AlphaFold and NMR, Structure (2022).↩︎
David Silver, Thomas Hubert, Julian Schrittwieser et al, Mastering Chess and Shogi by Self-Play with a General Reinforcement Learning Algorithm (2017).↩︎
David Silver, Thomas Hubert, Julian Schrittwieser et al, "A General Reinforcement Learning Algorithm That Masters Chess, Shogi, and Go Through Self-Play", Science (2018).↩︎
Natasha Regan and Matthew Sadler, "Game Changer: AlphaZero’s Groundbreaking Chess Strategies and the Promise of AI" (2019).↩︎
Julio González-Díaz and Ignacio Palacios-Huerta, AlphaZero Ideas (2022).↩︎
All of this is for classical games.↩︎
The original work was done by Neel Nanda and Tom Lieberum, and presented in A Mechanistic Interpretability Analysis of Grokking (2022). A fuller writeup may be found in: Neel Nanda, Lawrence Chan, Tom Lieberum, Jess Smith, and Jacob Steinhardt, Progress measures for grokking via mechanistic interpretability (2023).↩︎